
実験をしていると原因不明の失敗は度々あります。方法や試薬を変えてみてもうまく行かず、何度か繰り返すうちいつの間にか成功して、一体何が原因だったんだと思うこともよくあります。
おそらく方法や試薬ではなく手技の上達が問題を解決したのかも知れませんが、こういうのは永遠の謎です。
一方で原因不明の成功はあまり経験がありません。ところが先日久々にT-ベクターをつくったところ、やたらといいものができたのでちょっと紹介します。
このブログでも作成法を紹介したことがありますが(参考リンク→T-Vectorを使ったサブクローニング)、T-ベクターとは両切断端にTの3'突出がついた直鎖状プラスミドベクターのことです。
Taqポリメラーゼで増幅したDNA産物は3'末端にA突出があるのでライゲーション反応を行うとT-ベクターのT突出と相補的に会合してプラスミドとPCR産物がうまくくっついてくれます。
T-ベクター作成のおおまかな流れはこんな感じです。
(1)ベクターを平滑末端を生成する制限酵素で切断
(2)切断されたベクターを電気泳動で分離、精製
(3)デオキシチミジンの付加
(4)精製して分注、保存
今回もいつもと同じようにつくってライゲーションしてみたところ・・・
 |
| コロニーの生え方はこんな感じだが、 |
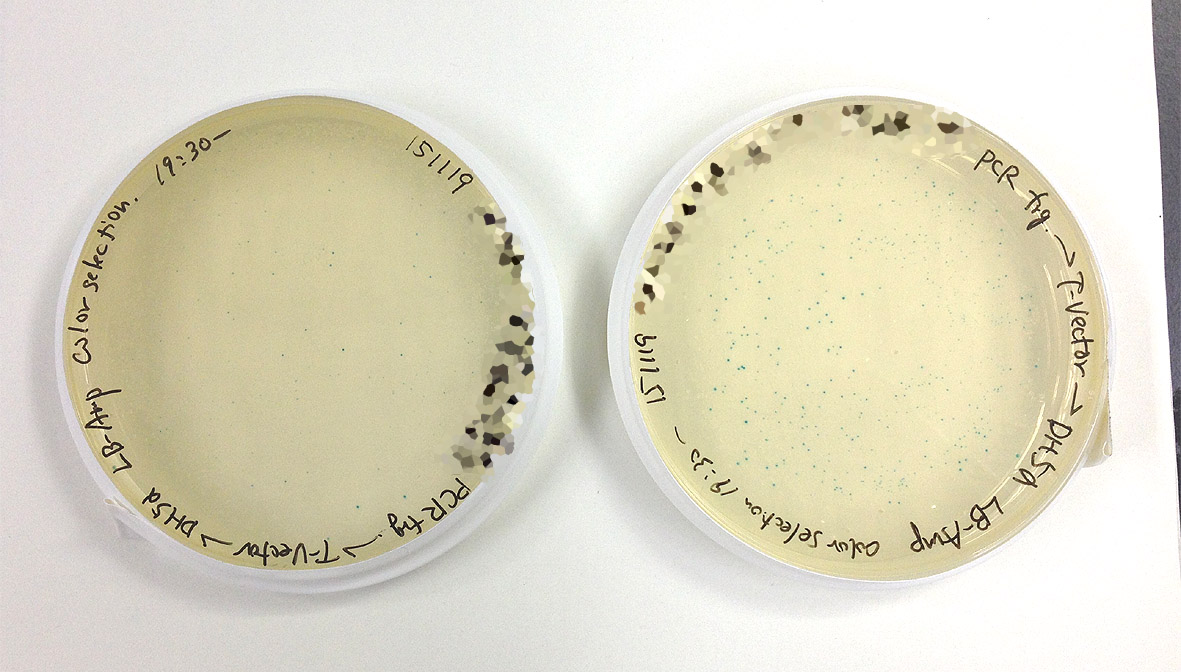 |
| 青コロニーがかなり少ない(画像クリックで拡大)。 |
特に左側のプレートにいたってはインキュベーターから出して見た瞬間、前日にX-galを入れ忘れたかと思ったほどの青コロニーの少なさでした。

 |
| そして白コロニーを32個拾ってチェックすると、全て入っている!(参考リンク→コロニーダイレクトPCR) |
こんなに効率のよいT-ベクターができたのは初めてです。たまたま今回まぐれでうまくいった可能性もありますが、せっかくなので作り方を記しておきます。以前紹介した方法と少しだけ違います。
(スーパー)T-Vectorの作成法
(1)ベクターの切断
pBluescriptII-SK+を使用しました。制限酵素はEcoRV。
| 1.5mLチューブに以下を混合 | 単位はμL | ||
| pBluescriptII SK+ (1μg/μL) | 2 | (計2μg) | |
| 10xBuffer H (TAKARA) | 10 | ||
| ddH2O | 85 | ||
| EcoRV | 3 | ||
| Total | 100 |
37℃ オーバーナイト処理しました。(おそらく5時間程度で良いと思います。)
(2)ベクターの精製
マッハライナーゲル社(TAKARA社) のNucleoSpinカラムキットで精製
 |
| このブログでよく使われるNucleoSpinカラムキット。 |
| 上記のチューブにBuffer NTを200μL加えて混ぜる | ||
| NucleoSpinカラムに溶液を移す。 | ||
| 遠心ろ過。液受けには付属2mLチューブを使用。 | 11,000xg 1min. | |
| ろ過された液は捨てる。 | ||
| Buffer NT3 (EtOH入リ) 750μLをカラムに加え遠心ろ過。 | 11,000xg 1min. | |
| カラムのメンブレンを乾かすため遠心処理 。 | 11,000xg 2min. | |
| ろ過された溶液は捨てる。 | ||
| カラムをDNA回収用の1.5mLチューブに移す。 | ||
| Buffer NEを15μL添加 | 室温2分放置 | |
| そのまま遠心分離でDNA溶液を回収 | 11,000xg 2min. |
つぎに電気泳動を行い、切断されたベクターを分離、ゲルの切り出しと精製を行います。
電気泳動にはミューピッドのミニゲル (0.7%) を使用し、大きい方のウェル型でゲルを作ります。バッファーは1xTAEを使います。
上記の過程で溶出したベクター溶液に色素を混ぜて全量を1つのウェルにアプライします。
100Vで30分電気泳動します。以前の方法では50V、1時間を推奨していましたが時間短縮のため変更しました。
泳動を止め、新しい染色液(染色タッパーに50mLの1xTAEと5μLのGelRedを混合)で20分染色します。
染色液を取り除き、ゲルをサランラップなどのラップの上に移してトランスイルミネーターへ。
 |
| またまた出ました、ゲルカッター。とても便利。 |
ゲル切り出しにはFastGENE社(日本ジェネティクス社)のゲルカッターを使用します。これも当ブログでよく出るアイテムです。前にも書きましたが便利な上に、何故かいつもキャンペーン中なのでおすすめです。
イルミネーターで確認しながら切断後直鎖状になったDNAのバンドを切り出し、1.5mLチューブに回収します。
ここでもまたNucleoSpinカラムでDNAを抽出、精製します。
 |
| NucleoSpinカラムと付属のチューブ |
| 回収したゲル断片に250μLのBuffer NTを添加 | ||
| 50℃で10分間放置。1000rpmで振盪または2-3分毎にボルテックスで撹拌 | ||
| NucleoSpinカラムに移し遠心ろ過。溶液受けには付属2mLチューブを使用。 | 11,000xg 1min. | |
| ろ過された溶液は捨てる。 | ||
| Buffer NT3 (エタノール入リ)750μLをカラムに加え遠心ろ過 | 11,000xg 1min. | |
| カラムのメンブレンを乾かすために何も入れず遠心処理 | 11,000xg 2min. | |
| カラムをDNA回収用の1.5mLチューブに移しBuffer NEを15μL添加 | 室温2分放置 | |
| そのまま遠心分離してDNA溶液を回収 | 11,000xg 2min. | |
(3)デオキシチミジンの付加
ここで使う試薬: 100mM dTTP (デオキシチミジン、TAKARA社)、Taqポリメラーゼ (NEB社)
 |
| デオキシチミジン (dTTP) |
| PCRチューブに以下の溶液を混合。(単位はμL) | ||
| 精製ベクター | 20 | |
| 10x Thermo Pol Buffer (NEB社 ポリメラーゼに添付) | 10 | |
| 100mM dTTP | 2 | |
| ddH2O | 67 | |
| NEB Taq polymerase | 1 | |
| Total | 100 | |
| サーマルサイクラーで以下の反応を行う。 | ||
| 68℃ 2時間 | ||
| 10℃ (4℃でもよい) hold |
(4)精製と分注、保存
しつこいようだがここでもやはりNucleoSpinカラムで精製する。
「私の研究室にはNucleoSpinカラムが常備されています。」
(Der NucleoSpin Säule hat in meinem Laboratorium gestanden.)
 |
| 本日3度めの登場。NucleoSpinカラム。 |
| 上記のチューブにBuffer NTを200μL加えて混ぜる | ||
| NucleoSpinカラムに溶液を移す。 | ||
| 遠心ろ過。液受けには付属2mLチューブを使用。 | 11,000xg 1min. | |
| ろ過された液は捨てる。 | ||
| Buffer NT3 (エタノール入リ) 750μLをカラムに加え遠心ろ過。 | 11,000xg 1min. | |
| ろ過された溶液は捨てる。 | ||
| もう一度Buffer NT3 750μLをカラムに加え遠心ろ過。 | 11,000xg 1min. | |
| カラムのメンブレンを乾かすため何も入れず遠心処理 。 | 11,000xg 2min. | |
| カラムをDNA回収用の1.5mLチューブに移す。 | ||
| 10mM Tris-HCl (pH8.0) を20μL添加 | 室温2分放置 | |
| そのまま遠心分離でDNA溶液を回収 | 11,000xg 2min. |
精製したT-ベクターを1~2μLずつPCRチューブに小分けして、-20℃で冷凍保存します。
以上で(super) T-vectorの作成は終了です。使用時に解凍して0.5-1μLを反応に用います。あまり凍結融解を繰り返さないようにしています。
【補足】 TAクローニング
PCR増幅産物はベクターと同様の方法で電気泳動でDNAバンドを切り出し、NucleoSpinで精製後10mM Tris-HCl溶液 10μL程度に溶かしておきます(参考リンク→ゲルからのDNAの切り出し)。
以下のPremix溶液をあらかじめ1.5mLチューブに作っておきます(例:2反応分)。
| 2x Quick Ligation Reation Buffer (NEB) | 5 | |
| T4 Ligase | 0.5 | |
| Total | 5.5μL |
僕はよくNEBのT4 Ligaseを使うのですが、今回の反応ではニッポンジーンの酵素を使いました(以前買ったものがあまっていた)。バッファーと酵素が別々のメーカーになりますが、問題ありません。
また、2x Quick Ligation Reaction BufferはNEB社のQuick Ligaseを買うと付いてくるバッファーですが、すでに使いきってしまったのでバッファーのみを購入して使っています → これ (リンク1) (リンク2)。
T4 Ligaseについてくるバッファーよりもこちらを使ったほうが圧倒的に効率がいいです。
 |
| ライゲーションは酵素やなか! バッファーたいっ! |
購入できるバッファーは組成が少しだけ違うものの同様に使えます。5x で売っているので購入後、水で薄めて2x Bufferにして冷凍保存 (-20℃) して使っています。
PCRチューブに以下を混合(1.5mLチューブでもいいと思いますが、溶液が少ないので蒸発に注意)。
| PCR増幅産物 | 2 | |
| T-Vector | 0.5 | |
| Premix溶液 | 2.5 | |
| Total | 5μL |
室温で15-20分間ライゲーション反応を行います。長時間の反応は避けます。
(Quick Ligation Reaction Bufferを使っているので、この後の熱変性は厳禁です。)
氷上で2分程度冷やした後、大腸菌にトランスフォームしプレートに撒きます。
長くなりましたが、効率が良いT-ベクターはとても重宝するので一度試してみてください。
No comments:
Post a Comment